

IBM AND DAIMLER USE QUANTUM COMPUTER TO DEVELOP NEXT-GEN BATTERIES
Electric vehicles have an Achilles heel: the capacity and speed-of-charging of their batteries. A quantum computing breakthrough by researchers at IBM and Daimler AG, the parent company of Mercedes-Benz, could help tackle this challenge. We used a quantum computer to model the dipole moment of three lithium-containing molecules, which brings us one step closer the next-generation lithium sulfur (Li-S) batteries that would be more powerful, longer lasting and cheaper than today’s widely used lithium ion batteries.
Simulating molecules is extremely difficult but modeling them precisely is crucial to discover new drugs and materials. In the research paper “Quantum Chemistry Simulations of Dominant Products in Lithium-Sulfur Batteries,” we simulated the ground state energies and the dipole moments of the molecules that could form in lithium-sulfur batteries during operation: lithium hydride (LiH), hydrogen sulfide (H2S), lithium hydrogen sulfide (LiSH), and the desired product, lithium sulfide (Li2S). In addition, and for the first time ever on quantum hardware, we demonstrated that we can calculate the dipole moment for LiH using 4 qubits on IBM Q Valencia, a premium-access 5-qubit quantum computer.
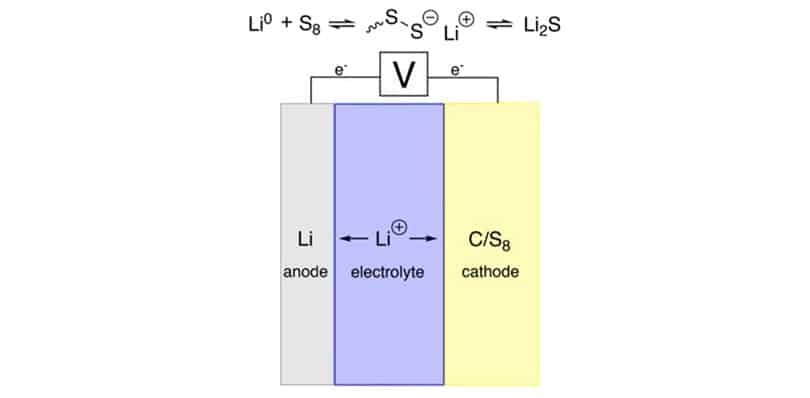
FIG. 1. The chemistry of a lithium-sulfur battery. During discharge,elemental sulfur is reduced into sulfur salts containing chains of vary-ing lengths, ultimately resulting in lithium sulfide production. Dur-ing charge, the chemistry is reversed. The mechanism for lithiumsulfide production is proposed to occur through a two-electron pro-cess.
Researchers at Daimler hope that quantum computers will help them design next-generation lithium-sulfur batteries, because they have the potential to compute and precisely simulate their fundamental behavior. Quantum computers are not yet better than classical computers. They are very ‘noisy,’ meaning that any outside disturbance knocks the fragile qubits out of quantum states crucial for the calculation too early for them to run meaningful computations. Still, they are already showing great promise in chemistry, towards precisely simulating complex molecules.
To get these results, we first evaluated the energies and dipole moments of all four molecules as we varied the atomic distance between the different atoms in the molecule (breaking the chemical bonds) using Qiskit. We then used the IBM Q Valencia device to calculate the dipole moments of LiH (and simulated the other three), a calculation to understand the electronic distribution in a molecule across its different atoms as bonds are broken. This is crucial – for instance, in the case of LiH, the dipole changes as the nuclei are separated, making the molecule change from being polar (ionic) to neutral. Also, while the molecular bonds are stretched, the molecular system must be described by a highly “entangled” state; entanglement is a property of quantum mechanics, according to which when one particle changes its state, so does its entangled ‘partner,’ simultaneously. The impact of this entanglement is visible in the dissociation curve for LiH, and without applying the error mitigation techniques, developed by IBM, there is a noticeable bump in the middle of the curve at around ~2.5 angstroms. We have applied the same error mitigation techniques in our recent work on Li–S batteries.
To make sure our calculations on the hardware were accurate, we also performed them on a classical computer using the IBM quantum simulator. Then, we ran these calculations on IBM Q Valencia, and compared the results. Despite working with noisy qubits, we were still able to extract sufficiently precise results.
What’s quantum chemistry?
The main aim of molecular simulation, on any machine, is to find a compound’s ground state—its most stable configuration. This is no trivial task because it requires simulating the interactions between all the particles, such as electrons, in the molecule. And the bigger and more complex a molecule and its environment is, the more difficult this process becomes.
Today’s supercomputers can simulate fairly simple molecules, but when researchers try to develop novel, complex compounds for better batteries and life-saving drugs, traditional computers can no longer maintain the accuracy they have at smaller scales. The solution has typically been to model experimental observations from the lab and then test the theory.
The largest chemical problems researchers have been so far able to simulate classically, meaning on a standard computer, by exact diagonalization (or FCI, full configuration interaction) comprise around 22 electrons and 22 orbitals, the size of an active space in the pentacene molecule. For reference, a single FCI iteration for pentacene takes ~1.17 hours on ~4096 processors and a full calculation would be expected to take around nine days. For any larger chemical problem, exact calculations become prohibitively slow and memory-consuming, so that approximation schemes need to be introduced in classical simulations, which are not guaranteed to be accurate and affordable for all chemical problems. It’s important to note that reasonably accurate approximations to classical FCI approaches also continue to evolve and is an active area of research, so we can expect that accurate approximations to classical FCI calculations will also continue to improve over time.
That’s where quantum computers come in. Qubits themselves operate according to the laws of quantum mechanics, just like the molecules researchers are trying to simulate. The hope is that in time quantum computers can greatly speed up the simulation process by precisely predicting the properties of a new molecule that can explain its behavior, such as reactivity. Programming qubits works by using unique properties of superposition and entanglement, allowing the potential for researchers to evaluate a expectation parameters – in a much more efficient way than a standard computer ever could.
In September 2017, a paper by an IBM team titled ‘Hardware-efficient Variational Quantum Eigensolver for Small Molecules and Quantum Magnets,’ on simulating hydrogen (H2), lithium hydride (LiH), and beryllium hydride (BeH2) molecules, made it onto the cover of Nature magazine. The research described a new hardware efficient ansatz to calculate the ground state of these molecules, by mapping the electronic structure of the orbitals onto a subset of a quantum processor, encoding from orbitals to qubits. The results were groundbreaking and laid the foundations of simulating different molecules with quantum computers.
Building on that, our latest calculations are an important step forward – as we keep expanding quantum computers’ capabilities to simulate the energies of larger and larger molecules with the same high levels of accuracy as small molecules. Just like our colleagues in their 2017 work, we also used the variational quantum eigensolver (VQE) algorithm to simulate elements of Li-S batteries. VQE is a hybrid classical-quantum algorithm that works by combining a classical and a quantum component.
As we improve the state of qubits, we’ll increase quantum volume, and the machines will become exponentially more powerful. So while we haven’t yet achieved quantum advantage, this type of research is the foundational work that will eventually get us there.
Author: Jeannette Garcia - Senior Manager for Quantum Applications, Algorithms and Theory, IBM Research












